


 购物车
购物车 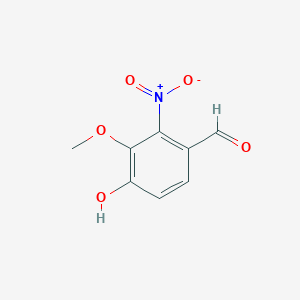
-
2-硝基-3-甲氧基-4-羟基苯甲醛
- names:
4-HYDROXY-2-NITRO-M-ANISALDEHYDE
- CAS号:
2450-26-2
MDL Number: MFCD00017020 - MF(分子式): C8H7NO5 MW(分子量): 197
- EINECS: Reaxys Number:
- Pubchem ID:96959 Brand:BIOFOUNT
| 货品编码 | 规格 | 纯度 | 价格 (¥) | 现价(¥) | 特价(¥) | 库存描述 | 数量 | 总计 (¥) |
|---|
| 中文别名 | 2-硝基-3-甲氧基-4-羟基苯甲醛(2450-26-2) |
| 英文别名 | 4-HYDROXY-2-NITRO-M-ANISALDEHYDE;4-Hydroxy-3-Methoxy-2-Nitrobenzaldehyde; 2450-26-2; 2-Nitrovanillin; Benzaldehyde, 4-Hydroxy-3-Methoxy-2-Nitro-; 4-Hydroxy-3-Methoxy-2-Nitro-Benzaldehyde; |
| CAS号 | 2450-26-2 |
| SMILES | COC1=C(C=CC(=C1[N+](=O)[O-])C=O)O |
| Inchi | InChI=1S/C8H7NO5/c1-14-8-6(11)3-2-5(4-10)7(8)9(12)13/h2-4,11H,1H3 |
| InchiKey | PHCNQUJHXJQLQR-UHFFFAOYSA-N |
| 分子式 Formula | C8H7NO5 |
| 分子量 Molecular Weight | 197 |
| 闪点 FP | No data available |
| 熔点 Melting point | 137-138 °C |
| 沸点 Boiling point | 387.8±42.0 °C |
| Polarizability极化度 | No data available |
| 密度 Density | 1.456±0.06 g/cm3 |
| 蒸汽压 Vapor Pressure | No data available |
| 溶解度Solubility | |
| 性状 | solid power |
| 储藏条件 Storage conditions | Store at room temp |
2-硝基-3-甲氧基-4-羟基苯甲醛???????合成方法1:
| Yield | Reaction Conditions | Operation in experiment |
|---|---|---|
| 96.3% | With potassium carbonate In methanol at 20℃; for 2.00 h; Acidic aqueous solution | A mixture of 4-formyl-2-methoxy-3-nitrophenyl acetate 4.54g [(L9.] [OMMOL)] and potassium carbonate 5.24g (37.9mmol) in methanol [40ML] was stirred at room temperature for 2 hours. The reaction mixture was poured into [WATER,-ACIDIFIED] by 1N [HC1] solution and extracted into AcOEt. The organic layer was washed with brine, dried over [MGS04,] filtrated and the solvent was evaporated. The residue was washed with n-hexane to give the title compound 3. [60G] as white solid. Yield 96.3percent. |
| 95% | Stage #1: With sodium hydroxide In water at 10 - 45℃; Stage #2: at 20 - 30℃; for 0.25 h; |
24 g acetyl-2-nitrovanilline were suspended in 300 ml water at 10-30 The mixture was heated to 40 - 45 and 40 g NaOH 30percent (300 mmol, 3 equiv.) were added in 10-20 min. Stirring was continued for 30-45 min. The reaction was cooled to 25-30 and the pH was set to 2.0-2.5 by the addition of 10percent sulfuric acid. Stirring was continued for 15 min at 20-25 and the product was filtered off and washed with water (2 x 100 ml). The product was dried at 45-50 under reduced pres sure and 1 8.7 g 2- nitrovanilline were obtained a white to yellow solid in 95percent yield. |
| 88% | Stage #1: at 20℃; for 16.00 h; |
Step 2: Preparation of 4-hvdroxy-3-methoxy-2-nitrobenzaldehvde; A mixture of 4-formyl-2-methoxy-3-nitrophenyl acetate 438 g (1.8 mol) and potassium carbonate (506 g, 3.7 mol) in MeOH (4000 mL) was stirred at room temperature for 16 h. ="67">The reaction mixture was concentrated under reduced pressure to afford a viscous oil. This was dissolved in water, acidified using a solution of HCI (2 N) and extracted with EtOAc. The organic layer was washed with brine, dried (MgSO4) and filtered. The solvent was concentrated under reduced pressure to 1/3 volume and the resulting solids were filtered and air-dried to give the title compound (317 g, 88percent): 1H NMR (DMSO-Cy6) δ: 9.69 (1 H1 s), 7.68 (1 H, d), 7.19 (1 H, d), 3.82 (3H, s). |
| 产品说明 | 2-硝基-3-甲氧基-4-羟基苯甲醛(2450-26-2)作为中间体使用,2450-26-2溶解度,熔点见主页 |
| Introduction | 4-HYDROXY-2-NITRO-M-ANISALDEHYDE(2450-26-2) |
| Application1 | |
| Application2 | |
| Application3 |
| Yield | Reaction Conditions | Operation in experiment |
|---|---|---|
| 62% | Stage #1: With sulfuric acid; nitric acid In dichloromethane; water at 0 - 5℃; for 12.00 h; Microwave irradiation; Large scale Stage #2: With potassium carbonate In methanol; dichloromethane at 30℃; for 3.00 h; Large scale Stage #3: With hydrogenchloride In dichloromethane; water at 30℃; for 0.25 h; Large scale |
2-Nitrovanilin (2) was synthesized via a flow nitration of vanillin acetate (1 ) in a micro reactor. 3.94 kg of nitric acid (65 wpercent) were added to 5.87 kg of concentrated sulfuric acid at 0 (nitrating acid). 1 .5 kg of va nillin acetate were dissolved in 2.9 kg of dichloromethane (vanillin acetate solution). Both solutions reacted in a micro reactor with flow rates of app. 8.0 mL/min (nitrating acid) and app. 4.0 mL/min (vanillin acetate solution) at 5. The reaction mi xture was directly dosed into 8 kg of water at 3 . After 3h flow rates were increased to 10 mL/min (nitrating acid) and 5.0 mL/min (vanillin acetate solution). After additional 9 h the flow reaction was completed. The layers were separated at r.t., and the aqueous phase was extracted with 2 L of dichloromethane. The combined organic phases were washed with 2 L of saturated sodium bicarbonate, and then 0.8 L of water. The dichloromethane solution was concentrated in vacuum to app. 3 L, 3.9 L of methanol were added and app. the same volume was removed by distillation again. Additional 3.9 L of methanol were added, and the solution concentrated to a volume of app. 3.5 L. 1 .25 kg of methanol were added, followed by 2.26 kg of potassium carbonate. The mixture was stirred at 30 for 3h. 7.3 kg of dichl oromethane and 12.8 kg of aqueous hydrochloric acid (10 wpercent) were added at < 30 (pH 0.5 - 1 ). The mixture was stirred for 15 min, and the layers were separated. The organic layer was filtered, and the filter cake washed with 0.5 L of dichloromethane. The aqueous layer was extracted twice with 4.1 kg of dichloromethane. The combined organic layers were concentrated in vacuum to app. 4 L. 3.41 kg of toluene were added, and the mixture concentrated to a final volume of app. 4 L. The mixture was cooled to 0. After 90 min the suspension was filtered. T he collected solids were washed with cold toluene and dried to give 0.95 kg (62 percent). 1 H-NMR (400 MHz, de-DMSO): δ = 3.84 (s, 3H), 7.23 (d, 1 H), 7.73 (d, 1 H), 9.74 (s, 1 H), 1 1 .82 (brs, 1 H). |
| Ca. 10 %Spectr. | Stage #1: With sulfuric acid; nitric acid In dichloromethane; water at 3 - 5℃; for 12.00 h; Flow reactor; Large scale Stage #2: at 30℃; for 3.00 h; Large scale |
Example 1 : Step A1 : Preparation of 4-acetoxy-3-methoxy-2- nitrobenzaldehyde (2) 3.94 kg of nitric acid (65 wpercent) were added to 5.87 kg of concentrated sulfuric acid at 0°C (nitrating acid). 1 .5 kg of vanillin acetate were dissolved in 2.9 kg of dichloromethane (vanillin acetate solution). Both solutions reacted in a micro reactor with flow rates of app. 8.0 mL/min (nitrating acid) and app. 4.0 mL/min (vanillin acetate solution) at 5°C. The reaction mixture was directly dosed into 8 kg of water at 3°C. After 3h flow rates were increased to 10 mL/min (nitrating acid) and 5.0 mL/min (vanillin acetate solution). After additional 9 h the flow reaction was completed. The layers were separated at r.t., and the aqueous phase was extracted with 2 L of dichloromethane. The combined organic phases were washed with 2 L of saturated sodium bicarbonate, and then 0.8 L of water. The dichloromethane solution was concentrated in vacuum to app. 3 L, 3.9 L of methanol were added and app. the same volume was removed by distillation again. Additional 3.9 L of methanol were added, and the solution concentrated to a volume of app. 3.5 L. This solution of 4-acetoxy-3-methoxy-2- nitrobenzaldehyde (2) was directly used in the next step. Example 2 : Step A2 : Preparation of 4-hydroxy -3-methoxy-2- nitrobenzaldehyde (2-nitro-vanillin) (3) To the solution of 4-acetoxy-3-methoxy-2-nitrobenzaldehyde (2) prepared as described in example 1 (see above) 1 .25 kg of methanol were added, followed by 2.26 kg of potassium carbonate. The mixture was stirred at 30°C for 3h. 7.3 kg of dichloromethane and 12.8 kg of aqueous hydrochloric acid (10 wpercent) were added at < 30°C (pH 0.5 - 1 ). The mixture was stirred for 15 min, and the layers were separated. The organic layer was filtered, and the filter cake washed with 0.5 L of dichloromethane. The aqueous layer was extracted twice with 4.1 kg of dichloromethane. The combined organic layers were concentrated in vacuum to app. 4 L. 3.41 kg of toluene were added, and the mixture concentrated to a final volume of app. 4 L. The mixture was cooled to 0°C. After 90 min the suspension was filtered. The collected solids were washed with cold toluene and dried to give 0.95 kg (62 percent). 1H-NMR (400 MHz, de-DMSO): δ =3.84 (s, 3H), 7.23 (d, 1 H), 7.73 (d, 1 H), 9.74 (s, 1 H), 1 1 .82 (brs, 1 H). NMR spectrum also contains signals of regioisomer 6-nitrovanillin (app. 10percent): δ = 3.95 (s, 3H), 7.37 (s, 1 H), 7.51 (s, 1 H), 10.16 (s, 1 H), 1 1 .1 1 (brs, 1 H). |
| 7.6 g | Stage #1: at -10 - 0℃; for 3.00 h; Stage #2: for 0.25 h; Reflux |
After fuming nitric acid (50 mL) was cooled at -20 ° C.,The acetate salt obtained above was added in small portions. Then,After stirring the mixture at -10 to 0 ° C. for 3 hours,Poured into ice cold water (~ 200 mL) and stirred for 10 minutes.The mixture was extracted with ethyl acetate, the combined organic layers were washed with brine,After drying with anhydrous Na 2 SO 4,Evaporation to dryness gave a crude product.The crude product obtained above was added to a 2 M KOH solution (200 mL)Reflux heating was carried out for 15 minutes.After cooling to room temperature, the reaction was stopped by slowly adding concentrated hydrochloric acid at 0 ° C.After filtering the solid matter, washing with cold water and vacuum drying,A mixture of Compound 1 as a brown solid and its regioisomer was obtained (7.6 g, |
[2] Patent: WO2016/71435, 2016, A2. Location in patent: Page/Page column 47-48
[3] Patent: JP2018/8900, 2018, A. Location in patent: Paragraph 0081; 0082; 0084; 0085
| 警示图 | |
| 危险性 | |
| 危险性警示 | No data available |
| 安全声明 | H317+H301 |
| 安全防护 | P261, P264, P270, P272, P280, P301+ |
| 备注 |
| Pictogram(s) |   |
|---|---|
| Signal | Danger |
| GHS Hazard Statements |
H301 (97.5%): Toxic if swallowed [Danger Acute toxicity, oral] H317 (97.5%): May cause an allergic skin reaction [Warning Sensitization, Skin] Information may vary between notifications depending on impurities, additives, and other factors. The percentage value in parenthesis indicates the notified classification ratio from companies that provide hazard codes. Only hazard codes with percentage values above 10% are shown. |
| Precautionary Statement Codes |
P261, P264, P270, P272, P280, P301 |
| First Total Synthesis of 9-Hydroxy-8H-pyrano[3,2-f]indol-2-one J. Heterocyclic Chem. 2015 10.1002/jhet.2251 |
| (E)-4-[(3,5-Dimethyl-phen-yl)imino-meth-yl]-2-meth-oxy-3-nitro-phenol;Acta crystallographica. Section E, Structure reports online;PMID:22259502 2012-01-01 |
| RSC Advances, 2017, vol. 7, # 82, p. 52180 - 52186 |
| Patent: CN107074876, 2017, A. Location in patent: Paragraph 0309; 0310; 0313 |
| Patent: JP5876570, 2016, B2. Location in patent: Paragraph 0407-0411 |
[1] Journal of Heterocyclic Chemistry, 2015, vol. 52, # 5, p. 1406 - 1410
[2] Patent: WO2004/29055, 2004, A1. Location in patent: Page 51
[3] Journal of Chemical Research, Miniprint, 1990, # 12, p. 2831 - 2867
[4] Patent: WO2016/71380, 2016, A1. Location in patent: Page/Page column 43
[5] Patent: WO2008/70150, 2008, A1. Location in patent: Page/Page column 65-66
[6] Patent: WO2012/62748, 2012, A1. Location in patent: Page/Page column 56
[7] Patent: WO2012/62743, 2012, A1. Location in patent: Page/Page column 60
[8] Patent: WO2012/62745, 2012, A1. Location in patent: Page/Page column 61
[9] ChemMedChem, 2016, p. 1517 - 1530
[10] Patent: US2015/266845, 2015, A1. Location in patent: Page/Page column 0051
[11] Patent: KR101770302, 2017, B1. Location in patent: Paragraph 0201; 0202
[12] Journal of Organic Chemistry, 1961, vol. 26, p. 4344 - 4347
[13] Chemical and pharmaceutical bulletin, 1965, vol. 13, # 2, p. 130 - 135
[14] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1993, # 17, p. 1967 - 1972
[15] Synthetic Communications, 1995, vol. 25, # 4, p. 569 - 572
[16] Journal of Physical Organic Chemistry, 2000, vol. 13, # 9, p. 511 - 517
[17] Tetrahedron Letters, 2000, vol. 41, # 49, p. 9369 - 9372
[18] Tetrahedron, 2002, vol. 58, # 17, p. 3423 - 3444
[19] Journal of Medicinal Chemistry, 2005, vol. 48, # 14, p. 4663 - 4669
[20] Organic Letters, 2006, vol. 8, # 14, p. 3117 - 3120
[21] Patent: US4287341, 1981, A
[22] Tetrahedron, 2009, vol. 65, # 16, p. 3246 - 3260
[23] Medicinal Chemistry Research, 2014, vol. 23, # 6, p. 2985 - 2994
[24] Journal of the American Chemical Society, 2015, vol. 137, # 19, p. 6219 - 6225
[25] Patent: WO2016/71382, 2016, A1. Location in patent: Page/Page column 41
- 相关产品
-
< >
- 推荐产品
-
< >
- 最新产品
-
< >
新闻
怎么做细胞爬片免疫组化染色实验
细胞爬片免疫组化染色,是通过细胞爬片是让玻片浸在细胞培养基内,细胞在玻片上生长,主要用于组织学,免疫组织化学...
2020/7/20 22:04:33
提取病毒RNA的实验方法
提取病毒RNA方法分别有:异硫氰酸胍的提取病毒RNA方法、TRIzol LS提取法、Trizol法提取法等等...
2020/7/22 20:29:26

各种微流控芯片键合方法的优缺点
微流控芯片键合:目前主要有激光焊接、热压键合、胶键合、超音波焊接,每种方法都有各自的优缺点。本文主要介绍聚酯...
2023/7/28 10:43:09

新一代微流控键合解决方案
微流控键合解决方案:微流控芯片制造的一个重要环节,也是最容易被忽视的--芯片键合。其中一个重要因素是:微流控...
2023/7/27 12:44:28
荧光素钾盐使用说明
D-荧光素钾盐(K+)设计用于体外和体内生物发光测定。D-荧光素的质量和纯度对于获得良好和可重复的结果至关重...
2023/7/20 11:05:11
如何选BSA(牛血清白蛋白)
如何选BSA(牛血清白蛋白):牛血清白蛋白(BSA)有多种形式,如何选择适合自己的牛血清白蛋白(BSA)是一...
2023/2/14 13:09:18

牛血清白蛋白(BSA)常见问题
牛血清白蛋白(BSA)常见问题:牛血清白蛋白(BSA)在实验室中是通用的,可用于蛋白质印迹、细胞组织培养、P...
2022/10/19 9:39:51
pubmed使用方法(技巧)
pubmed使用方法(技巧):PubMed是一个关于医学问题的学术文章和书籍的数据库。因为它是一份学术期刊,...
2022/10/18 18:06:07
BSA(牛血清白蛋白)
BSA(牛血清白蛋白):牛血清白蛋白(BSA)是一种球状蛋白质,牛血清白蛋白(BSA)是发现于牛血浆中的主要...
2022/10/18 16:48:12
冻干培养细菌的方法
冻干培养细菌的方法:冷冻干燥,也称为冻干或冷冻干燥,是在产品冷冻后除去水分并将其置于真空中的过程。这使得冰可...
2022/10/16 8:27:31



