

 购物车
购物车 T7 DNA聚合酶测序技术(实验简介,实验步骤,注意事项,实验所需试剂)
Release Time:2020-07-31一、T7 DNA聚合酶测序技术实验简介:
T7 DNA聚合酶测序实验中,T7 DNA聚合酶是第一批同时拥有两种活性的转移酶类,分别是5'→3'聚合酶活性与单链及双链3'→5'核酸外切酶活性,因此如果预先使用适当的方法处理T7 DNA聚合酶,则实验中的3'→5'核酸外切酶的活性也会随之大大降低,此时经处理后修饰完成的T7 DNA聚合酶也称为T7测序酶。通过使用T7测序酶获得的测序数据在每个碱基位置具有相对均匀的配对参与。因此,通过放射自显影获得的结果相对清晰并且易于辨别。
T7测序酶具有高聚合速度和高度连续性。因此,该酶在使用中不同于Klenow片段和逆转录酶。它几乎没有错误终止,整个反应可在短时间内完成,并且不需要其他反应(追赶步骤)。然而,对于具有紧密二级结构的区域,错误的终止反应将导致读取序列的困难。如果遇到这些情况,则应使用某些高温DNA聚合酶,例如Promega的测序级Taq DNA酶。利用高温DNA聚合酶独特的耐热性,测序反应可以在不利于二级结构形成的高温条件下进行。
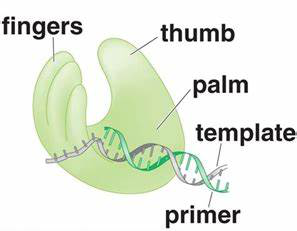
二、T7 DNA聚合酶测序技术实验步骤
(一)模板准备
1. M13单链模板的制备:
转化为合适的大肠杆菌宿主菌株并接种在含有指示剂(例如X-gal / IPTG)的中上后,含有M13重组体的细胞将显示出无色的“噬菌斑”,实际上已被感染。细胞不会被溶解或杀死通过噬菌体。出现噬菌斑是因为被感染的细菌比周围未感染的细菌生长慢。从无色噬菌斑获得的感染细胞可通过培养测序反应所需的单链模板生产。
(1)将培养过夜的宿主细胞(例如NM522或JM101)用3ml TYP肉汁中1:100稀释。在37°C剧烈振荡1小时后,细胞进入早期对数期。此时,用滴管将含有重组M13(与琼脂)的合适噬菌斑转移到细胞中中。
(2)在37°C下剧烈振摇(300 rpm)孵育6小时。
(3)将细胞中转移到两个1.5ml 离心管中,并以12000g离心15分钟。将上清液转移到新的离心管中并离心15分钟。小心取出1-1.2ml上清液(注意不要触摸沉淀物),然后转移到新的离心管中。
(4)在上清液中加入0.25体积的3.75M醋酸铵(pH 7.5),其中含有20%的PEG(-8000)以沉淀噬菌体。混合后,将其置于冰上30分钟,然后以12000g离心15分钟,倒出上清液,然后以相同的方式再次离心。用移液器吸头完全吸收剩余的PEG。此时,试管底部应有芝麻大小的噬菌体颗粒沉淀物。
(5)将沉淀物重新溶解在400ml TE缓冲液中。
(6)加入等体积的氯仿/异戊醇(24:1)混合物,剧烈摇动1分钟,然后以12000g离心5分钟。
(7)将水相转移到新的离心管中,注意不要触摸原始管中的两相界面。加入等体积的苯酚/氯仿(1:1)混合物,剧烈摇动1分钟,然后以12000g离心5分钟。
(8)将上层水相转移到另一个新的离心管中,重复步骤7的萃取,必要时重复几次,直到两相之间没有可见的物质。
(9)将上层水相转移至新的离心管中,加入等体积的氯仿,剧烈摇动1分钟,以12000g离心5分钟,然后重复此步骤数次。
(10)将上层水相转移到新的离心管中,加入7.5 mol / L乙酸铵的0.5倍体积,乙醇的2倍体积,混合均匀并在-20°C放置30分钟。
(11)以12000g离心15分钟,除去上清液,用70%乙醇小心洗涤沉淀物,如果沉淀物已经搅拌,请重新离心,倒出酒精,并真空干燥沉淀物。
(12)将DNA沉淀溶于20ml去离子水中,取2ml样品进行琼脂糖凝胶电泳定量,如果DNA量足够,则进行退火测序。
2.噬菌粒单链模板的制备:
噬菌粒是指含有丝状单链DNA噬菌体复制区的嵌合质粒。分子克隆中常用的pBluescript系列和pGEM系列质粒均为噬菌粒。含有重组噬菌粒的单菌落细胞在液体中培养并用辅助噬菌体超感染后,即可产生用于测序反应的单链DNA模板。
(1)从新鲜培养皿中挑选出一个含有pGEM质粒DNA的抗Amp菌落,并将其接种到100ml含50mg / ml氨苄青霉素的TYP肉汤中,并在37℃振摇过夜。
(2)将100ml过夜培养物接种到另一个装有5ml TYP肉汁中(含50mg / ml Amp)的新试管中,并在37°C剧烈振摇30分钟。
(3)加入40μl辅助噬菌体R408或M13 K07,继续剧烈搅拌并振摇6-8小时,以使感染性噬菌体超感染。
(4)以12000g离心15分钟后,除去沉淀物,取上清液,转移到新的离心管中,再次离心15分钟,小心地取1?1.2ml上清液(注意不要触摸沉淀物)。
(5)在上清液中加入0.25体积的3.75M醋酸铵,其中含有20%的PEG,以沉淀噬菌体颗粒。混合后,将其置于冰上30分钟,然后以12000g离心15分钟,倒出上清液,然后以相同的方式再次离心。使用移液器吸头吸收剩余的PEG溶液。此时,您应该看到芝麻大小的噬菌体颗粒在试管底部沉淀。
(6)将沉淀物溶于400ml TE缓冲液(10mmol / L Tris-HCl pH8.0,1mmol / L EDTA)中。
(7)加入等体积的氯仿/异戊醇(24:1)混合物,剧烈摇动1分钟,然后以12000g离心5分钟。
(8)将水相转移至干净的离心管中(不要触摸原始管中两相的界面),加入等体积的苯酚/氯仿(1:1)混合,剧烈摇动1分钟,并以12000g离心5分钟。
(9)将上层水相转移到干净的离心管中,并重复第八步萃取,如有必要,重复几次,直到两相之间没有可见的物质。
(10)将上层水相转移到干净的离心管中,加入等体积的氯仿,剧烈摇动1分钟,然后离心,重复此步骤数次。
(11)将上层水相转移至干净的离心管中,加入7.5 mol / L pH 7.5醋酸铵的0.5倍体积,然后加入2倍体积的乙醇。混合后,将其在-20°C下放置30分钟。
(12)以12000g离心15分钟,除去上清液,用70%乙醇小心洗涤沉淀物,如果已搅拌沉淀物,则再次离心,倒出乙醇,并真空干燥沉淀物。
(13)将沉淀物溶于20ml去离子水中,取2ml样品,然后进行琼脂糖凝胶电泳进行定量。如果DNA量足够,请执行退火和测序。
(二)超螺旋质粒DNA的碱性变性
1.将4mg(约2pmol)超螺旋质粒DNA放入离心管中,加去离子水至最终体积18ml。
2.加入2 ml 2 mmol / L NaOH / 2 mol / L EDTA溶液,并在室温(25℃)下放置5分钟。
3.加入2ml 2mol / L NaAc溶液(pH 4.6)并涡旋混合均匀以中和。
4.加入75ml的无水乙醇,充分混合,然后放在干冰或-70°C下放置10分钟。
5.以12000g离心10分钟,弃去上清液,用200ml预冷的70%乙醇洗涤沉淀,干燥沉淀,并将其溶于20ml去离子水中。
(三)测序反应
1.底漆和模板的退火:
(1)在离心管中混合以下试剂:
①底漆约1?2pmol,②5×T7 DNA聚合酶测序反应缓冲液2ml,③DNA约1mg,④将ddH2O加入最终体积至10ml。
(2)将试管加盖后,在65°C加热2分钟,然后在大约30分钟内将试管的温度缓慢降至室温。在冷却过程中,可以使用小型加热板或小型保温仪器,也可以使用已调节至65°C的水浴,这样它们的温度才能在室温下缓慢降至室温。 。当温度降至30°C以下时,退火结束。可以将小试管放在冰上,并且必须在4小时内使用退火的模板。
2.标记反应
(1)在标准反应过程中(从引物开始,可读到500多个碱基),首先需要将标记混合物以1:5的比例稀释。对于4毫升混合物,加入16毫升双蒸馏去离子水。稀释后的溶液在-20°C下可以使用数周。如果要确定的序列少于30个碱基,则可以将标记缓冲液稀释15倍,并且模板DNA应当高于0.5pmol。由于DNA模板的量不足,标记反应的程度将降低,即仅标记引物后的几个核苷酸。
(2)用冰冷的TE缓冲液稀释T7 DNA聚合酶1:8。稀释后应立即使用酶溶液,并且不应将其置于冰浴中超过60分钟。请勿使用标记混合物,DTT溶液或非缓冲型溶液稀释酶溶液。
(3)在退火的底漆-模板混合物中,添加:①0.1mol/L DTT 1.0ml,②标记液2.0ml,③[a-35P] dATP或[a-35S] dATP 0.5毫升,④2.0ml稀释的T7 DNA聚合酶,⑤充分混合(注意不要产生气泡),并在室温下放置5-10分钟。,⑥如果在电泳过程中出现谱带压缩问题,则dITP混合物的效果要好于dGTP混合物,并且dITP混合物的使用与dGTP混合物的使用相似。
3.链终止反应:
(1)取4个离心管,并用G,A,T,C标记。
(2)在每支试管中加入2.5ml G,A,T,C链端终止混合物,并立即关闭盖子以防止液体蒸发(此反应最好在标记反应之前完成)。根据需要选择dGTP和dITP的混合物。
(3)将离心管预热至37°C 1分钟。
(4)标记反应完成后,将3.5ml标记反应溶液放入4个离心管中,离心一点点,然后将4个管置于37°C孵育。请注意,在每个反应中,都应使用新的移液器吸头以避免交叉污染。
(5)保持3至5分钟(如果使用dITP,则保持时间可以延长至30分钟,而对反应产物没有任何影响)。
(6)在上述四个试管中分别加入4ml终止缓冲液。充分混合后,置于冰浴中。可以加载该样品进行电泳。标有35S的样品可以在-20°C下保存1周,这时样品几乎不会降解。为避免降解,需要在同一天对32P标记的电泳。
(四)测序凝胶板的制备及电泳
有关测序凝胶板的制备和电泳,请参见第一部分。将样品加热到75?80℃2分钟,然后立即上样。每个泳道的使用量为2?3ml。
(五)测序胶板的干燥和放射自显影
电泳后,可以通过对b射线敏感的X射线胶片放射自显影法检测标记为32P或35S的产物。
1.取下电泳玻璃板,除去两侧的珠子,然后用一块小的薄钢板撬开玻璃板。如果玻璃板用键合硅烷处理,则凝胶将紧紧粘附在玻璃板上,并且凝胶将在以下步骤中与玻璃板一起加工。如果玻璃板未经过硅烷键合处理,则可以使用3MM大滤纸在玻璃板上粘贴凝胶,小心地剥离凝胶,然后准备进行下一步处理。
2.要除去尿素,请将装有凝胶的玻璃板或滤纸浸入装有2升10%冰醋酸的大型展示盆中,然后轻轻摇动15分钟以除去尿素。
3.干胶:可以使用电动吹风机干燥包含凝胶的玻璃板,而可以使用专用的干燥胶设备(例如真空加热干胶计)将包含凝胶的滤纸排干。
4.干燥的凝胶可用于放射自显影。将X射线胶片添加到装有凝胶的玻璃板的侧面后,用另一块类似尺寸的玻璃板夹住它,然后用夹子固定,并将其放在暗盒中进行曝光。可以将包含凝胶的滤纸放在X射线曝光箱中以进行放射自显影。通常,32P暴露是过夜,而35S是2到3天。
5.曝光后,可以对X射线胶片进行处理以获得序列信息。首先将X射线胶片放在水中以使其润湿,然后将其放在显影液中显影,直到清晰地显示条带为止。用水冲洗X射线胶片一段时间,然后将其放入定影液中定影15分钟以上。取出X射线胶片以干燥并读取序列。
三、T7 DNA聚合酶测序技术实验注意事项:
(一)标记反应中:
1.稀释时,应在加入酶溶液之前将所有溶液完全混合。
2.在标记反应的最后一步骤的过程中,如果反应过程中发生多次冷却,保持时间过长或温度过高,则测序反应会短于100个碱基。
(二)测序胶板的干燥和放射自显影过程中:
1.尿素的清除非常重要。如果残留尿素,凝胶在干燥过程中将非常粘,无法进行放射自显影。可以用10%冰醋酸洗涤两次,第一次15分钟,第二次10分钟。如果胶水浓度高于12%,则在干燥过程中可能会出现皱纹。有两种预防方法:(1)在冰醋酸溶液中添加1%至2%的甘油; (2)请勿擦干胶水。直接在冰箱中以冷冻状态暴露。应该注意的是,当使用这种方法时,应在凝胶和X射线胶片之间添加一层塑料薄膜。一般而言,使用第一种方法。
2.凝胶在曝光前必须充分干燥,否则X射线胶片会粘在凝胶表面,从而使后续操作变得困难。
3.暴露时间应根据所用同位素确定。如果同位素的半衰期已过,则应相应延长暴露时间。
四、T7 DNA聚合酶测序技术实验所需试剂清单:
| 序列 | 产品名 | 货号 | CAS | 备注 |
| 1 | Tris-HCl | HC2034 | ||
| 2 | MgCl2 | JT2113 | 7786-30-3 | |
| 3 | NaCl | JT0001 | 7647-14-5 | |
| 4 | DTT(二硫苏糖醇) | HCM000375 | 3483-12-3 | |
| 5 | 2′-脱氧鸟苷 5′-三磷酸 三钠盐(DGTP) | SS0814 | 93919-41-6 | |
| 6 | 三磷酸脱氧胞苷钠盐(DCTP) | SS0956 | 102783-51-7 | |
| 7 | 2'-脱氧胸苷-5'-三磷酸三钠,二水(DTTP) | SS0808 | 18423-43-3 | |
| 8 | BSA | JT0982 | ||
| 9 | 甲酰胺 | HXH504144 | 75-12-7 | |
| 10 | EDTA | BSH83093 | 162303-59-5 | |
| 11 | 溴酚蓝 | SX0029 | 115-39-9 | |
| 12 | 二甲苯蓝 | SX0100 | 4463-44-9 | |
| 13 | 无水亚硫酸钠(无水Na2SO3) | SS0124 | 7757-83-7 | |
| 14 | 对苯二酚 | JT24022 | 123-31-9 | |
| 15 | 无水碳酸钠 | JT8648 | 497-19-8 | |
| 16 | KBr | JT0893 | 7758-02-3 | |
| 17 | 36%乙酸 | JT11990 | 64-19-7 | |
| 18 | 硼酸 | JT0008 | 10043-35-3 | |
| 19 | K2HPO4 | SS1346 | 16788-57-1 | |
| 20 | PEG500溶液 | HC1100 | ||
| 21 | 2ml圆底连盖离心 | F-EP020 | ||
| 22 | 1.5ml尖底连盖离心管 | F-EP015 |



